南湖新闻网讯(通讯员 赵伦 周强伟)近日,我校博中bz会员登录李国亮教授、植物科学技术学院赵伦教授联合南方科技大学朱健康团队在Genome Biology发表题为DNA methylation underpins the epigenomic landscape regulating genome transcription in Arabidopsis的研究论文。该研究利用DNA甲基化完全缺失的拟南芥五重突变体mddcc(met1 drm1 drm2 cmt2 cmt3)等材料、高效的eChIP-seq等技术和新开发的DNA甲基化分析方法,深入解析了DNA甲基化与组蛋白修饰的协作关系及其在基因组转录调控中的作用,获取了一系列新认识。
DNA甲基化在动植物中非常保守,与组蛋白修饰等表观修饰共同塑造染色质状态,在基因转录、基因组稳定性和生长发育等过程中发挥重要作用。由于缺乏完全无DNA甲基化的材料和相关技术不足,人们对DNA甲基化与组蛋白修饰间的协作关系及其功能的认知受到限制。
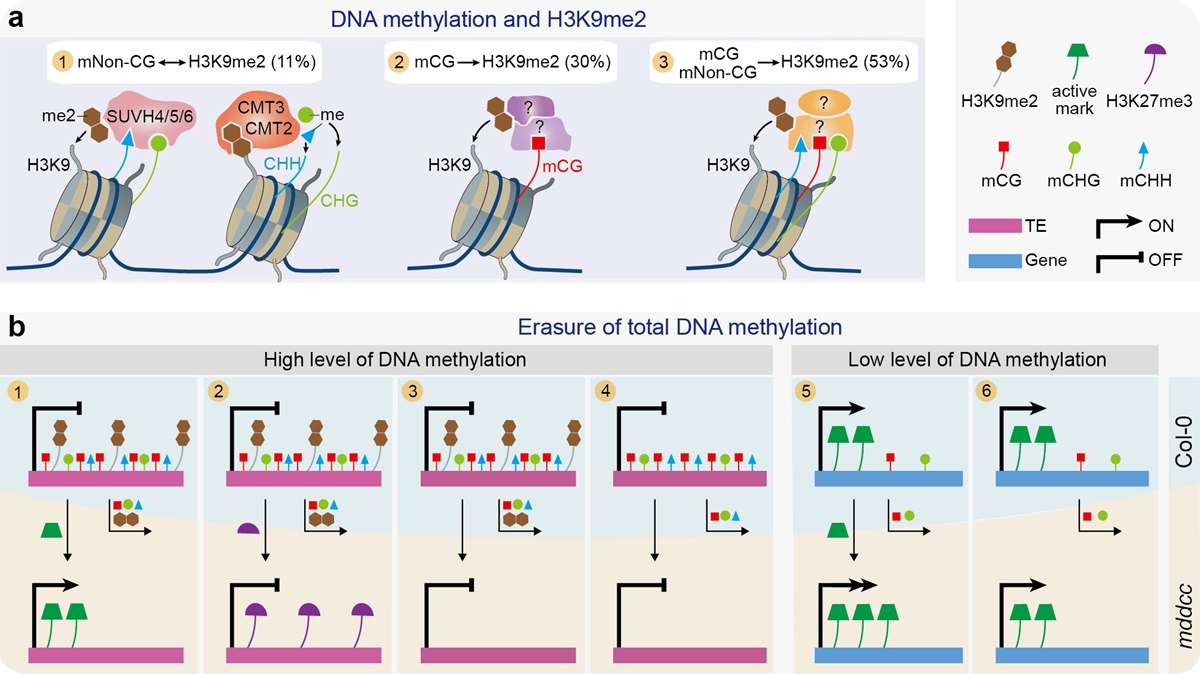
胞嘧啶残基C-5位的DNA甲基化是一种保守的表观遗传修饰。在植物中,DNA甲基化发生在CG、CHG和CHH(H表示A、T或C)三种碱基序列的胞嘧啶上。CG甲基化由DNA甲基转移酶MET1维持,CHG甲基化由CMT3和CMT2维持,CHH甲基化则由CMT2和DRM1/2维持。尽管这五种DNA甲基转移酶的作用机制已被广泛研究,但对它们是否足以维持整个基因组的 DNA甲基化尚不清楚。前期,朱健康团队首次创建了DNA甲基化完全缺失突变体mddcc。mddcc表现出生长迟缓、极端矮小、不能开花结实等一系列严重的发育缺陷,因此材料极难获得;而细胞学和形态学等方面的研究表明DNA甲基化对多个植物发育过程至关重要。mddcc材料的成功创建为研究DNA甲基化与组蛋白修饰等表观遗传标记间的协作调控及其在特定生物过程的作用提供了材料基础。
由于只能获得极少量的mddcc材料,难以利用常规ChIP-seq技术开展表观基因组学研究。前期,赵伦博士主导开发了一种快速、高效的植物ChIP-seq方法eChIP-seq。与植物常用ChIP-seq方法相比,eChIP-seq的总体材料使用效率提升至少几十倍以上,可用极少量植物材料鉴定组蛋白修饰和转录因子全基因组结合位点,为该研究提供了技术支撑。
该研究综合利用三种DNA甲基化缺失突变体(mddcc:无DNA甲基化;met1:无CG甲基化;ddcc:无non-CG甲基化)和eChIP-seq技术,结合李国亮团队开发的DNA甲基化分析方法,深入解析了DNA甲基化与不同类型组蛋白修饰间的协作关系及其在基因组转录调控中的作用。
团队系统研究了DNA甲基化在组蛋白修饰/染色质状态转变中的调控作用。在上述三种DNA甲基化突变体中产生了超过120套的表观基因组学数据,包括6种代表性组蛋白修饰(H3K4me3, H3K9ac, H3K27ac, H3K4me1, H3K27me3和H3K9me2)、DNA甲基化组和转录组等,为研究提供了重要资源。研究发现,DNA甲基化完全消失造成约35%的组蛋白修饰水平和分布发生变化,揭示了DNA甲基化在调控染色质状态变化中的重要作用。更进一步,该研究确定了不同类型表观修饰对基因组不同位点和转录活性的优先调控顺序,定量评估了不同类型DNA甲基化(CG甲基化和non-CG甲基化)在染色质状态调控/转化中的作用。
该研究发现,只有在mddcc中H3K9me2才全部消失,证实全基因组H3K9me2的沉积依赖于DNA甲基化的存在。进一步研究表明,CG甲基化特异调控约30%的H3K9me2位点,而non-CG甲基化特异调控约11%的H3K9me2位点,说明CG甲基化在H3K9me2调控中发挥更大的作用。再者,一部分位点的H3K9me2修饰由CG甲基化和non-CG甲基化独立控制,而另一部分位点修饰(约53%)是由两者协同调控的。这些发现革新了对于DNA甲基化与H3K9me2相互协作的现有认识。
研究还在植物中首次证实活跃组蛋白修饰和多梳抑制性标记H3K27me3是一种不依赖于DNA甲基化的转录调控系统。在DNA甲基化完全缺失突变体mddcc中,异染色质标记H3K9me2消失,但依然存在(保留或获得)多种其他类型的组蛋白修饰,主要包括活跃(H3K4me3, H3K9ac, H3K27ac和H3K4me1)和多梳抑制性(H3K27me3)等修饰。该研究发现mddcc中存在的这些组蛋白修饰依然表现出与其在野生型中类似的转录调控功能。因此,研究者认为,在植物中活跃组蛋白修饰和多梳抑制性标记H3K27me3是一种不依赖于DNA甲基化的转录调控系统。这个惊人的观点得到了来自无DNA甲基化生物(比如秀丽隐杆线虫)的佐证。在一些生物中,没有检测到DNA甲基化修饰,但存在多种组蛋白修饰,这些组蛋白修饰发挥了重要的转录调控功能。
研究发现H3K27me3可能是一种DNA甲基化/H3K9me2丢失后维持转座子沉默的替补机制。表明植物进化出DNA甲基化、H3K9me2和H3K27me3等多种转座子沉默机制,以保证基因组的稳定性。
该研究不仅为植物表观遗传/基因组学相关研究提供了重要数据资源,而且深入揭示了DNA甲基化在染色质表观修饰可塑性的贡献,并为复杂的表观调控网络在基因组转录调控中的作用提供了新认知。
南方科技大学朱健康教授和我校李国亮教授为共同通讯作者,我校赵伦教授(原朱健康团队博士后)、周强伟博士以及中科院上海植物逆境生物学研究中心何力副研究员为共同第一作者。华中农业大学邓利博士和德国图宾根大学Rosa Lozano-Duran教授等参与了该研究。
审核人:李国亮 赵伦
【英文摘要】
It is challenging to determine the effect of DNA methylation on the epigenetic landscape and the function in higher organisms due to the lack of DNA methylation-free mutants.
Here, the analysis of a recently generated Arabidopsis mutant completely devoid of DNA methylation reveals that DNA methylation underpins the genome-wide landscape of histone modifications. Complete loss of DNA methylation causes an upheaval of the histone modification landscape, including complete loss of H3K9me2 and widespread redistribution of active and H3K27me3 histone marks, mostly owing to the role of DNA methylation in initiating H3K9me2 deposition and excluding active marks and repressive mark H3K27me3; CG and non-CG methylation can act independently at some genomic regions while they act cooperatively at many other regions. The transcriptional reprogramming upon loss of all DNA methylation correlates with the extensive redistribution or switches of the examined histone modifications. Histone modifications retained or gained in the DNA methylation-free mutant serve as DNA methylation-independent transcriptional regulatory signals: active marks promote genome transcription, whereas the repressive mark H3K27me3 compensates for the lack of DNA hypermethylation/H3K9me2 at multiple transposon families.
Our results show that an intact DNA methylome constitutes the scaffolding of the epigenomic landscape in Arabidopsis and is critical for controlled genome transcription and ultimately for proper growth and development.
论文链接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02768-x